- Stable download
- Development code
- General notes
- MS-DOS notes
- Unix/Linux notes
- Compilation
- Using the command line
- Viewing output files
- Version history
- Running PLINK
- PED files
- MAP files
- Transposed filesets
- Long-format filesets
- Binary PED files
- Alternate phenotypes
- Covariate files
- Cluster files
- Set files
- Recode
- Reorder
- Write SNP list
- Update SNP map
- Update allele information
- Force reference allele
- Update individuals
- Write covariate files
- Write cluster files
- Flip strand
- Scan for strand problem
- Merge two files
- Merge multiple files
- Extract SNPs
- Remove SNPs
- Zero out sets of genotypes
- Extract Individuals
- Remove Individuals
- Filter Individuals
- Attribute filters
- Create a set file
- Tabulate SNPs by sets
- SNP quality scores
- Genotypic quality scores
- Missingness
- Obligatory missingness
- IBM clustering
- Missingness by phenotype
- Missingness by genotype
- Hardy-Weinberg
- Allele frequencies
- LD-based SNP pruning
- Mendel errors
- Sex check
- Pedigree errors
- IBS clustering
- Permutation test
- Clustering options
- IBS matrix
- Multidimensional scaling
- Outlier detection
- Case/control
- Fisher's exact
- Full model
- Stratified analysis
- Tests of heterogeneity
- Hotelling's T(2) test
- Quantitative trait
- Quantitative trait means
- Quantitative trait GxE
- Linear and logistic models
- Set-based tests
- Multiple-test correction
- Basic permutation
- Adaptive permutation
- max(T) permutation
- Ranked permutation
- Gene-dropping
- Within-cluster
- Permuted phenotypes files
- Imputing haplotypes
- Precomputed lists
- Haplotype frequencies
- Haplotype-based association
- Haplotype-based GLM tests
- Haplotype-based TDT
- Haplotype imputation
- Individual phases
- Making reference set
- Basic association test
- Modifying parameters
- Imputing discrete calls
- Verbose output options
- File format
- MAP file construction
- Loading CNVs
- Check for overlap
- Filter on type
- Filter on genes
- Filter on frequency
- Burden analysis
- Geneset enrichment
- Mapping loci
- Regional tests
- Quantitative traits
- Write CNV lists
- Write gene lists
- Grouping CNVs
- Overview/example
- Basic usage
- Consistency checks
- Aliases
- Joint IDs
- Lookups
- Replace values
- Match files
- Quick match files
- Misc.
- HapMap (PLINK format)
- Teaching materials
- Multimarker tests
- Gene-set lists
- Gene range lists
- SNP attributes
Rare copy number variant (CNV) data
This page describes some basic file formats, convenience functions and analysis options for rare copy number variant (CNV) data. Support for common copy number polymorphisms (CNPs) is described here. Copy number variants are represented as segments. These segments are essentially represented and analysed in a similar manner to how PLINK handles runs of homozygosity (defined by a start and stop site on a given chromosome). Allelic (i.e. basic SNP) information is not considered here: PLINK skips the usual procedure of reading in SNP genotype data. Here we assume that some other software package such as the Birdsuite package has previously been used to make calls for either specific copy-number variable genotypes or to identify particular genomic regions in individuals that are deletions or duplications, based on the raw data. That is, PLINK only offers functions for downstream analysis of CNV data, not for identifying CNVs in the first place, i.e. similar to the distinction between SNP genotype calling versus the subsequent analysis of those calls. In this section, we describe the basic format for rare CNV data; the steps involved in making a MAP file and loading the data. We consider ways to filter the CNV lists by type, genomic location or frequency. We describe options for relating CNVs to phenotype, either at the level of genome-wide burden or looking for specific associations. Finally, we detail the tools for producing reports of any genes intersected by CNVs and for displaying groups of overlapping CNVs.Basic support for segmental CNV data
The basic command for reading a list of segmental CN variants is
plink --cnv-list mydata.cnv
--fam mydata.fam
--map mydata.cnv.map
which can be abbreviated
plink --cfile mydata
(note that the map file must have the .cnv.map map extension). The CNV list file mydata.cnv has the format
FID Family ID
IID Individual ID
CHR Chromosome
BP1 Start position (base-pair)
BP2 End position (base-pair)
TYPE Type of variant, e.g. 0,1 or 3,4 copies
SCORE Confidence score associated with variant
SITES Number of probes in the variant
Having a header row is optional; if the first line starts with FID
it will be ignored.
Note The SCORE and SITES values
are not used in any direct way, except potentially as variates to filter
segments on, as described below. That is, the values of these do not
fundamentally impact the way analysis is performed by PLINK
itself (they might alter the meaning of the results of course, e.g. if
including low-confidence calls into the analysis!). In other words, if
whatever software was used to generate the CNV calls does not supply
some conceptually similar values, it is okay to simply put dummy codes
(e.g. all 0) in these two fields.
The first few lines of a small example file is shown here:
FID IID CHR BP1 BP2 TYPE SCORE SITE
P1 P1 4 71338469 71459318 1 27 0
P1 P1 5 31250352 32213542 1 34.2 0
P1 P1 7 53205351 53481230 3 18.2 0
P2 P2 11 86736484 87074601 1 22 0
P2 P2 14 47817280 47930190 4 55.1 0
...
The FAM file format is the first 6 fields of a PED file,
described here; this file lists the sex,
phenotype and founder status of each individual. The MAP file format
is described here, although the next
section how this can be automatically created using
the --cnv-make-map command.
Creating MAP files for CNV data
Prior to any analysis, a dummy MAP first needs to be created (this step only needs to be performed once per CNV file). This PLINK-generated MAP file has dummy entries that correspond to the start and stop sites of all segments. This facilitates subsequent parsing and analysis of CNV data by PLINK. The --cnv-make-map command is used as follows:plink --cnv-list mydata.cnv --cnv-make-map
which creates a file
plink.cnv.map
which will look just like a standard MAP file but with
dummy markers:
1 p1-51593 0 51593
1 p1-51598 0 51598
1 p1-51666 0 51666
1 p1-52282 0 52282
1 p1-69061 0 69061
...
where the marker names start with the p prefix and contain
chromosome and base-position information.
As an (unrealistic) example to illustrate how the mapping works,
consider the following, with 3 segments, spanning "positions" 1 to 8,
4 to 12 and 16 to 23. In this case, 6 unqiue map positions would be
created, the three start positions and the three stop positions.
Base 1111111111222222
Position 1234567890123456789012345
Marker # 1..2...3...4...5......6..
| | | | | |
| | |
Segments *------* | |
*-------*
*------*
The new MAP file would then be
1 p1-1 0 1
1 p1-4 0 4
1 p1-8 0 8
1 p1-12 0 12
1 p1-16 0 16
1 p1-23 0 23
Given such a MAP file, these three segments would then be perfectly
mapped to the corresponding markers (p1-1 to p1-8,
p1-4 to p1-12 and p1-16
to p1-23). The created MAP file is then specified in
subsequent segmental CNV analyses (using --cnv-list) with the
standard --map command (or --cfile command).
Loading CNV data files
Once a suitable MAP file has been created, i.e. with dummy markers that correspond to the position of every start and stop site of all segments, use the --cnv-list command again to load in the CNV segment data. As mentioned above, in addition to the basic CNV file, a MAP (previously generated) and FAM file (continaing ID and phenotype information) also need to be specified. For example.plink --map plink.cnv.map --fam mydata.fam --cnv-list mydata.cnv
Alternatively, if the MAP, FAM and CNV list files all have the same root, the commandplink --cfile study1
is equivalent, i.e. it implies the following files exist
study1.cnv
study1.cnv.map
study1.fam
By default either command will simply load in the CNV data and produce
a report in the LOG file, enumerating the number of CN states in the
total dataset and any filtering processes applied. For example,
Reading segment list (CNVs) from [ cnv1.list ]
714 of 2203 mapped as valid segments
1872 mapped to a person, of which 714 passed filters
CopyN Count
0 46
1 339
3 200
4 129
Writing segment summary to [ plink.cnv.indiv ]
This indicates that of 2203 total segments (i.e. should correspond to
number of lines in the cnv1.list file, allowing for any
header) 1872 are mapped to a person in the dataset. In other words,
some of the segments in cnv1.list are for individuals not in
cnv1.fam. These are simply ignored; for example, these
individuals might have been filtered out of the study for other
reasons, e.g. QC based on standard SNP genotypes. Of these, 714 passed
the further set of filters, as described below. As described below,
segments can be filtered based on genomic location, frequency, size,
quality score/number of sites and type (duplication or deletion).
It will also be reported in the LOG file if some of the segments do
not map to a marker in the MAP file: if this is because you've
used --chr or similar commands to restrict the portion of the
data examined, you can safely ignore this line; otherwise, it might
mean that the appropriate MAP file wasn't created
(e.g. using --cnv-make-map) for that CNV file.
By default, PLINK will create a file that summarises per
individual events (after any filtering has been applied), in a file
named
plink.cnv.indiv
which has the fields, one row per person, in the same order as the original FAM file:
FID Family ID
IID Individual ID
PHE Phenotype
NSEG Number of segments that individual has
KB Total kilobase distance spanned by segments
KBAVG Average segment size
PLINK will also create a file
plink.cnv.summary
that represents a count of CNVs, in cases (AFF) and controls (UNAFF)
that overlap each map position.
Checking for overlapping CNV calls (within the same individual)
As a sanity check of a CNV file: to check whether segments are overlapping for the same person (e.g. if a deletion and a duplication event had been specified for the same person in the same region, or if the same event is listed twice), use the optionplink --cfile mydata --cnv-check-no-overlap
If there is overlap, this writes a warning to the LOG, with the number of implicated events:
Within-individual CNV overlap detected, involving 2 CNVs
and creates a file
plink.cnv.overlap
that lists these offending segments, with the format:
FID Family ID
IID Individual ID
CHR Chromosome code
BP1 Segment start (bp)
BP2 Segment end (bp)
Filtering of CNV data based on CNV type
The segments read in can be filtered in a number of ways. First, one can specify to read in only either deletions (TYPE is less than 2) or duplications (TYPE is greater than 2), with the options,--cnv-deland
--cnv-dupSegments can also be filtered based on a minimum size (kb), score or number of sites contributing with the following commands:
--cnv-kb 50 --cnv-score 3 --cnv-sites 5The default minimum segment size is 20kb; none of the other filters have a default setting that would exclude anything. Also, corresponding maximum thresholds can be set:
--cnv-max-kb 2000 --cnv-max-score 10 --cnv-max-sites 10As mentioned above, the SCORE and SITES fields are not used for any other purpose in analysis, and so if you do not have this information, can can safely enter dummy information (e.g. a value of 1 for every CNV). The set of individuals for whom segment data are based on can be modified with the standard --keep and --remove options, to exclude people from the analysis.
Filtering of CNV data based on genomic location
It is possible to extract a specific set of segments that overlap with one or more regions as specified in a file, e.g. that might contain the genomic co-ordinates for genes or segmental duplications, etc. Use the command
--cnv-intersect regions.list
The file regions.list should be in the following format: one range per line, whitespace-separated:
CHR Chromosome code (1-22, X, Y, XY, MT, 0)
BP1 Start of range, physical position in base units
BP2 End of range, as above
MISC Any other fields after 3rd ignored
For example, if regions.list were
2 30000000 35000000 REGION1
2 60000000 62000000
X 10000000 20000000 Linkage hotspot
then
plink --cfile mydata --cnv-intersect regions.list
would extract all segments in mydata.cnv that at least partially span these three regions (5Mb and 2Mb on chromosome 2 and 10Mb on chromosome X), ignoring the comments or gene names. A typical type of file used with --cnv-intersect will often be a list of genes (such as available in the resources page). Alternatively, you can use
--cnv-exclude regions.list
to filter out a specific set of segments, i.e. to remove any CNVs that overlap with
one or more regions specified in the file regions.list.
Assuming the region file has consistent, unique names in the fourth field, the command
--cnv-subset mylist.txt
takes a list of region names and extracts just these from the
main --cnv-intersect, --cnv-exclude
(or --cnv-count, as described below) list. e.g. if mylist.txt contained
REGION1
REGION2
and region.list where
2 30000000 35000000 REGION1
2 60000000 62000000 GENE22
X 10000000 20000000 LinkageHotspot
then only the first region (chromosome 3, 30Mb to 35Mb, labelled
REGION1) would be extracted, as REGION2 does not exist. The --cnv-subset
command requires that the regions.list file has exactly four fields
(i.e. always a unique region/gene name in the fourth field).
Defining overlap for partially overlapping CNVs and regions
The basic intersection or exclusion commands will select all segments that are at least partially in the specified region. Alternatively, one can select only segments that have at least X percent of them in the specified region, for example
--cnv-overlap 0.50
would only include (--cnv-intersect), or exclude (--cnv-exclude),
events that have at least 50% of their length spanned by the region.
There are two other variant forms of the overlap command, which change
the denominator in calculating the proportion overlap:
--cnv-union-overlap 0.50
which defines overlap as the ratio of the intersection and the union,
also
--cnv-region-overlap 0.50
which defines overlap as the ratio of the intersection and the length
of the region (rather than the CNV). For example,
------|-----|--------------------- Region/gene
----------+++++++++++++++--------- CNV (duplication, +)
----------XXX--------------------- Intersection
----------XXXXXXXXXXXXXXX--------- Denominator for basic overlap
------XXXXXXXXXXXXXXXXXXX--------- Denominator for union overlap
------XXXXXXX--------------------- Denominator for region overlap
In this example, if we take each character to represent a standard length
Default overlap = 3 / 15
Union overlap = 3 / 19
Region overlap = 3 / 7
This next example illustrates how the overlap statistics can then
subsequently be used to include or exclude specific CNVs: if overlap
threshold were set to 0.5, then only the first of therse two CNVs would
be selected by --cnv-intersect
------|----------|----------------
--------OOOOOOOOOOXXX------------- Selected
------------OOOOOOXXXXXXXXXXXXXX-- Not selected
The default setting is equivalent to setting
--cnv-overlap 0 (i.e. more than 0% must
overlap).
Finally, the command
--cnv-disrupt
will select only CNVs that start or stop within a region
specified in the region list (i.e. resulting in a partially deleted or
duplicated gene or region). The normal overlap commands cannot be used
in conjunction with the --cnv-disrupt defintion of whether or
not a CNV overlaps a gene.
Filtering by chromosomal co-ordinates
In addition, the standard commands for filtering chromosomal positions are still applicable, for example
--chr 5
or
--chr 2 --from-mb 20 --to-mb 25
Note that for a CNV to be included when using these filters, both the start and stop site must fall within
the prespecified range (i.e. a CNV spanning from 19 to 24Mb on chromosome 2 would not be included in the above example).
Filtering of CNV data based on frequency
It is also possible to exclude based on the frequency of CNVs at a particular position. There are two main approaches to this: by assigning frequencies for regions and then applying the same routines as for the range-intersection command described above, or alternatively by assigning each CNV a single, specific count. These commands, and the differences between them, are described more fully on this page. As well as the two basic approaches described above, one can specify different degrees of overlap when calculating frequencies, which can alter the result of frequency filtering. The key commands and some examples are given here. To remove segments that map to regions with more than 10 segments
--cnv-freq-exclude-above 10
To remove any segments that only have at most 4 copies
--cnv-freq-exclude-below 5
To remove any segments not in regions with exactly 5 copies
--cnv-freq-exclude-exact 5
and correspondingly to include only segments in regions with exactly 5 copies
--cnv-freq-include-exact 5
As with the earlier range intersection commands, the definition
of intersection can be soft, specified with
the --cnv-overlap option. In most cases here, one would
probably want to allow for a soft filtering,
e.g. with --cnv-overlap 0.5 for example.
For example, given the following segments, and counts below
Segments *------*
*------------*
*------*
Counts 001112222211111112211111100000
Common regions XXXXX XX
then --cnv-freq-exclude-above 1 would remove all three
segments if --cnv-overlap 0 (the default) were set. This is
because each CNV has at least some part of it that intersects with a
region that contains more than 1 CNV. However,
if --cnv-overlap were instead set to 0.5, for example, then
only the top segment would be removed (as the other two segments have
more than 50% of their length outside of a region with more than 1
segment). If the overlap were set higher still, then in this example no
CNVs would be removed by the command --cnv-freq-exclude-above 1.
NOTE Because multiple CNVs at the same region will
not all exactly overlap, and may be spanned by distinct larger events,
or contain smaller events, in other individuals, then requesting that
you include only CNVs with exactly five copies for example
(--cnv-freq-include-exact 5) does not mean that at all
positions in the genome you will always see either 0 or 5
copies. Rather, the selection process works exactly as specified
above. Please see this page for further
details.
Alternative frequency filtering specification
The alternate approach is invoked with the command
--cnv-freq-method2 0.5
where the value following it represents an overlap parameter (there is no
need to specify the --cnv-overlap command directly when using
--cnv-freq-method2). Based on this overlap, PLINK will assign a specific
count to each CNV that represents the number of CNVs that overlap it (including itself)
based on a union intersection overlap definition with the specified proportion parameter,
between that CNV and all CNVs.
This approach is illustrated in the page, that gives more details
on the frequency filtering commands including a comparison to the region-based approach to filtering,
described above.
If the --cnv-freq-method2 command is used, then the other frequency filtering commands
will use the CNV-based counts to include of exclude CNVs, for example
plink --cfile mydata
--cnv-freq-method2 0.5
--cnv-freq-exclude-above 10
If --cnv-write (see below) is specified with --cnv-freq-method2,
then the additional command
--cnv-write-freq
will add a field FREQ to the plink.cnv file generated that shows the frequency for each CNV.
Also, the --cnv-seglist command (see below) can be modified with --cnv-write-freq
(to report the frequency as a number at the start and stop of each CNV instead of the usual codes).
Miscellaneous commands frequency filtering commands
To keep only segments that are unique to either cases or to controls
--cnv-unique
This can be used in conjunction with other frequency filter
commands. To drop individuals from the file who do not have at least
one segment after filtering, add the flag
--cnv-drop-no-segment
This can make the plink.cnv.indiv summary files easy to
browse, for example.
Burden analysis of segmental CNV data
To perform a set of global test of CNV burden in cases versus controls, add the
--cnv-indiv-perm
option as well as
--mperm 10000
for example (i.e. permutation is required). By default, this reports on four tests,
which use these metrics to calculate burden in both cases and controls
RATE Number of segments
PROP Proportion of sample with one or more segment
TOTKB Total kb length spanned
AVGKB Average segment size
Tests are based (1-sided) on comparing these metrics in cases versus controls,
evaluated by permutation.
If a list of regions is supplied in a file, e.g. gene.list and the command
--cnv-count gene.list
then an extra test is added
GRATE Number of regions/genes spanned by CNVs
GPROP Number of CNVs with at least one gene
GRICH Number of regions/genes per total CNV kb
These tests respect all the normal filtering commands, with the exception
that --cnv-intersect and --cnv-exclude cannot be used if
--cnv-count is also being used.
The mean metrics in cases and controls are reported in the file
plink.cnv.grp.summary
when the --cnv-indiv-perm command is used. For example: this gives
the number of events (N) in cases and controls, the rate per person,
the proportion of cases/controls to have at least one event, the total distance
spanned per person and the average event size per person.
TEST GRP AFF UNAFF
N ALL 528 362
RATE ALL 0.1557 0.1138
PROP ALL 0.1309 0.1041
TOTKB ALL 290.8 265.4
AVGKB ALL 249.8 243.3
As usual, if the --within command is added and a cluster file
specified, then any permutations are performed within cluster. In this
case, the statistics displayed in the plink.cnv.grp.summary
file are also split out by the strata as well as presented in total
(as indicated by the GRP field).
Improved gene-set enrichment analysis for segmental CNV data
This test implements a geneset-enrichment method for CNV data described in Raychaudhuri et al, 2010. It is appropriate for either continuous or disease traits and allows for the inclusion of multiple other covariates and for empirical significance tests.
The following examples illustrate basic usage. If the file genes.dat contains the locations of all genes (i.e. as available from here and the file pathway.txt is a file of gene names forming the pathway to be tested for enrichment and the CNV data are in the files mycnv.cnv, mycnv.cnv.map and mycnv.fam as described above, then one can ask whether a) genes are enriched for CNVs, b) a subset of genes are enriched, relative to the whole genome, c) a subset of genes are enriched, relative to all genes. The latter form of the enrichment test might be desirable, for example, to determine whether any enrichment is general to all genes, or specific to a subset of genes.
To test for enrichment of genic CNVs:
plink --cfile mycnv
--cnv-count genes.dat
--cnv-enrichment-test
Enrichment of pathway genes CNVs, relative to all CNVs
plink --cfile mycnv
--cnv-count genes.dat
--cnv-subset pathway.txt
--cnv-enrichment-test
To test for enrichment of pathway genes CNVs, relative to all genic CNVs
plink --cfile mycnv
--cnv-intersect genes.dat
--cnv-write my-genic-cnv
plink --cfile my-genic-cnv
--cnv-count genes.dat
--cnv-subset pathway.txt
--cnv-enrichment-test
The usual modifiers (to define intersection differently, allow for a certain kb border around each gene, filter on CNV size, type or frequency, etc) are all available. Under all circumstances, 2-sided asymptotic p-values are returned. Alternatively, permutation testing can be applied and 1-sided empirical p-values are returned (positive enrichment in cases, based on estimated regression coefficient), by adding the flag:
--mperm 10000
Association mapping with segmental CNV data
To perform a simple permutation-based test of association of segmental CNV data for case/control phenotypes, add the option
--mperm 50000
to perform, for example, 50,000 null permutations to generate empirical p-values. T
he results are saved in the file
plink.cnv.summary.mperm
This is a standard empirical p-value file: EMP1 and EMP2 represent pointwise
and genome-wide corrected p-values, respectively. Both tests are 1-sided by default.
You can consult the corresponding
plink.cnv.summary
that is also generated for details of the association: this file has the fields
CHR Chromosome code
SNP SNP identifier (dummy SNP, see below)
BP Base-pair position
AFF Number of affected individuals with a segment at this position
UNAFF Number of unaffected individuals
To instead perform a 2-sided test (i.e. allowing that events might be more common in controls) add
the flag
--cnv-test-2sided
To perform an analysis in which the total number of events within a
sliding window is compared between cases and controls (rather than the
number overlapping a single position) add the flag
--cnv-test-window 50
where the parameter is the kb window either side of the test
position. As before, the association results are reported per marker,
but now the counts indicate the total number of segments that overlap
any of the 100kb window surrounding the test position (+/- 50kb),
rather than just the test position itself. Significance is evaluated
by permutation as before.
Association mapping with segmental CNV data: regional tests
To perform a test of association for CNVs in particular regions, use the command./plink --cfile mydata --cnv-intersect glist-hg18 --cnv-test-region --mperm 10000
where glist-hg18 contains a list of genes (as available from the resources page. The output is written to
plink.cnv.regional.summary
which has the fields
CHR Chromosome code
REGION Name of region
BP1 Start position of region
BP2 End position of region
AFF Number of case CNVs spanning region
UNAFF Number of control CNVs spanning region
and the permutation results are written to
plink.cnv.regional.summary.mperm
which has the fields
CHR Chromosome code
REGION Name of region
STAT Statistic
EMP1 Empirical p-value, per region
EMP2 Empirical p-value, corrected for all tests
For example, the line
CHR REGION BP1 BP2 AFF UNAFF 1 TTLL10 1079148 1143176 2 3 ...implies 2 case CNVs (note, PLINK does not distinguish whether these CNVs belong to the same individual or not) and 3 control CNVs span the gene TTLL10. The standard commands for regions in CNV analysis such as --cnv-border and --cnv-overlap can be used in this context.
Association mapping with segmental CNV data: quantitative traits
To test for association between rare CNVs and a quantitative trait, use the same commands as for disease traits. PLINK will automatically detect that the phenotype is continuous. For example, if the file pheno.qt contains a quantitative trait, the command./plink --cfile mydata --pheno qt.dat --mperm 10000
will generate a file
plink.cnv.qt.summary
which contains the fields
CHR Chromosome code
SNP Dummy label for map position
BP Physical position (base-pairs)
NCNV Number of individiuals with a CNV here
M1 QT mean in individuls with a CNV here
M0 QT mean in individuals without a CNV here
and the file
plink.cnv.qt.summary.mperm
that contains the empirical p-values, EMP1 and EMP2,
as for disease traits. The only difference is that the quantitative
trait test is, by default, two-sided. To perform a 1-sided CNV test,
add the command
--cnv-test-1sided
NOTE Currently, genome-wide burden
(--cnv-indiv-perm), window-based (--cnv-test-window)
and region-based (--cnv-test-region) CNV association tests
are not available for quantitative traits.
Writing new CNV lists
Given a set of filters applied, you can output as a new CNV file the filtered subset, with the command
--cnv-write
For example, to make a new file using only deletions over 200kb but not more
than 1000kb, with a quality score of 10 or more, use the command
plink --cfile cnv1
--cnv-del
--cnv-kb 200
--cnv-max-kb 1000
--cnv-score 10
--cnv-write
--out hiqual-large-deletions
which will generate two new files
hiqual-large-deletions.cnv
hiqual-large-deletions.fam
To obtan a corresponding MAP file, so that you can subsequently use
--cfile hiqual-large-deletions
give the command
plink --cnv-list hiqual-large-deletions.cnv --cnv-make-map --out hiqual-large-deletions
(although note that this will overwrite the LOG file generated by the --cnv-write command).Creating UCSC browser CNV tracks
As opposed to listing CNVs in PLINK format with --cnv-write, the command --cnv-track will generate a UCSC-friendly BED file (note: this is distinct from a PLINK binary PED file) that can be uploaded to their browser for convenient viewing.plink --cfile mydata --cnv-track --out mycnvs
which generates a file
plink.cnv.bed
The filtering commands described above can be combined with this option.
By using the Manage custom tracks option on
the UCSC genome
browser, one can easily visualise the CNV data, along side other
genomic features. For example, the file (IID
and SCORE, SITES information is omitted for clarity)
FID IID CHR BP1 BP2 TYPE SCORE SITES
... ... 22 20140420 20241877 1 ... ...
... ... 22 20140420 20241877 1 ... ...
... ... 22 20129453 20241877 1 ... ...
... ... 22 20140609 20241877 1 ... ...
... ... 22 20140420 20241877 1 ... ...
... ... 22 20140420 20241877 1 ... ...
... ... 22 20639721 20793965 1 ... ...
... ... 22 20639721 20765489 1 ... ...
... ... 22 20305076 20591362 3 ... ...
... ... 22 20646213 20756780 3 ... ...
... ... 22 20140420 20259122 1 ... ...
... ... 22 20639866 20787533 3 ... ...
... ... 22 20140420 20241877 1 ... ...
... ... 22 20140420 20241877 1 ... ...
... ... 22 20140420 20241877 1 ... ...
... ... 22 20140420 20241877 1 ... ...
... ... 22 20348901 20498220 3 ... ...
... ... 22 20140420 20241877 1 ... ...
... ... 22 20140420 20241877 1 ... ...
... ... 22 20639643 20793173 3 ... ...
... ... 22 20140420 20241877 1 ... ...
... ... 22 20141114 20241877 1 ... ...
... ... 22 20140420 20254215 1 ... ...
... ... 22 20140420 20241877 1 ... ...
... ... 22 20129130 20241877 1 ... ...
is rendered
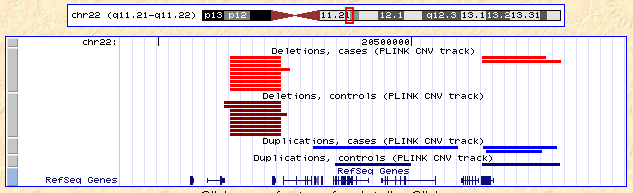 Note that the CNVs are split by deletion versus duplication (red versus blue) and case versus control (light versus dark).
Additionally, a poor-man's version of this plot can be obtained with the
command
Note that the CNVs are split by deletion versus duplication (red versus blue) and case versus control (light versus dark).
Additionally, a poor-man's version of this plot can be obtained with the
command
--cnv-seglist
which produces a file
plink.cnv.seglist
which, for the CNV list above, can be seen here. Deletions
and duplications are represented by + and - symbols at the start of each CNV; case
and control status is represented as A and U.
Finally, it is also possible to report CNVs annotated by the regions or genes
they span (see --cnv-verbose-report-regions
described below.
Listing intersected genes and regions
With the --cnv-intersect (or --cnv-exclude) command, you can add the flag
--cnv-report-regions
which will create a file
plink.reg
listing only the regions that intersect (or do not intersect) with any of the
CNVs (given the filtering and overlap commands that might also be specified). For example,
to obtain a list of genes that are intersected by a rare case singleton deletions over 500kb (i.e. event seen only once)
plink --cfile mydata
--filter-cases
--cnv-freq-exclude-above 1
--cnv-del
--cnv-kb 500
--cnv-report-regions
--cnv-intersect glist-hg18
Alternatively, the command
--cnv-verbose-report-regions
produces a verbose form of plink.reg, which does not just list the regions or genes intersected
but lists the specific segmental CNVs also. This can be used in conjunction with, for example,
--cnv-subset genes.txt
in order to produce reports on specific genes of interest. For example if genes.txt contained
HES4
ISG15
then
plink --cfile mydata
--cnv-verbose-report-regions
--cnv-intersect glist-hg18
--cnv-border 20
--cnv-subset genes.txt
would produce a file
plink.reg
that, for each gene/region, contains the following fields
FID Family ID
IID Individual ID
PHE Phenotype
CHR Chromosome code
BP1 Start position (base-pair)
BP2 Stop position (base-pair)
TYPE DELetion or DUPlication
KB Kilobase length of CNV
OLAP Overlap (extent of CNV covered by gene)
OLAP_U Union overlap (ration of intersection to union)
OLAP_R Region overlap (extent of gene covered by CNV)
that might contain something like the following report
RANGE (+/- 20kb ) [ 1 924206 925333 HES4 ]
FID IID PHE CHR BP1 BP2 TYPE KB OLAP OLAP_U OLAP_R
P001 1 2 1 789258 1232396 DUP 443.1 0.09281 0.09281 1
P002 1 1 1 826576 1304312 DEL 477.7 0.08609 0.08609 1
P003 1 2 1 864765 1913364 DUP 1049 0.03922 0.03922 1
P004 1 1 1 890974 1258710 DUP 367.7 0.1118 0.1118 1
RANGE (+/- 20kb ) [ 1 938709 939782 ISG15 ]
FID IID PHE CHR BP1 BP2 TYPE KB OLAP OLAP_U OLAP_R
P001 1 2 1 789258 1232396 DUP 443.1 0.09269 0.09269 1
P002 1 1 1 826576 1304312 DEL 477.7 0.08598 0.08598 1
P003 1 2 1 864765 1913364 DUP 1049 0.03917 0.03917 1
P004 1 1 1 890974 1258710 DUP 367.7 0.1117 0.1117 1
That is, this is a list of any CNV that at least partially overlaps
these two genes. The exact behavior can be modified with flags such
as --cnv-del, --cnv-kb, --cnv-disrupt, --cnv-overlap,
--filter-cases, etc.
Reporting sets of overlapping segmental CNVs
Finally, there are two option to group or report sets of segments that span a particular position. In the first case, use the option
--segment-group
which takes all segments in a given region (whole genome unless
otherwise specified) and forms "pools" of overlapping
segments. Several pools of overlapping segments will be created; these
will be listed in order of decreasing size (number of segments); note
that the same segment can appear in multiple pools (e.g. if A overlaps
with C, and B overlaps with C, but A and B do not overlap). The pools
give information as described below.
The more restricted form of this command forms a single pool of all
segments that overlap a particular position, which takes a single
parameter of a marker name; typically these will be the dummy pos*
markers created by the --cnv-make-map command.
--segment-spanning pos119
In this case, for some made-up data, we see from
the plink.cnv.summary file that there are 8 cases and 6
controls with a segment spanning a particular
position, pos586
CHR SNP BP AFF UNAFF
... ... ... ... ...
1 pos586 16631570 8 6
... ... ... ... ...
In this case, there is unsurprisingly no association between segmental
CNVs and disease: for example, the corresponding position in
the plink.cnv.summary.mperm file shows an empirical p-value
of 0.35, but of p=1 if adjusted for multiple testing (EMP2)
CHR SNP STAT EMP1 EMP2
... ... ... ... ...
1 pos586 0.419408 0.351324 1
... ... ... ... ...
Naturally, one would usually be more interested in following up significantly
associated regions of course... Nonetheless, if so desired we can see which
segments (given any of the filtering specified) are spanning this position, with
--segment-spanning, which gives the following:
POOL FID IID PHE CHR BP1 BP2 KB TYPE SCORE
S1 PT-2378 PT-2378 2 12 16631570 16751087 119.517 DEL 10.23
S1 PT-268D PT-268D 2 12 16631494 16732162 100.668 DEL 9.3
S1 PT-2M8O PT-2M8O 1 12 16631441 16751082 119.641 DEL 31.23
S1 PT-2FZ9 PT-2FZ9 2 12 16631436 16751045 119.609 DEL 15.2
S1 PT-287D PT-287D 1 12 16616579 17183201 566.622 DUP 200.3
S1 PT-2C91 PT-2C91 2 12 16616579 16751045 134.466 DEL 14.3
S1 PT-28A8 PT-28A8 1 12 16616579 16751045 134.466 DEL 8.3
S1 PT-2FPB PT-2FPB 1 12 16616579 16714372 97.793 DEL 11.1
S1 PT-28IG PT-28IG 2 12 16616579 16708856 92.277 DEL 10.3
S1 PT-2E5N PT-2E5N 2 12 16614664 16715703 101.039 DEL 9.87
S1 PT-2FVL PT-2FVL 1 12 16614664 16751045 136.381 DEL 10.67
S1 PT-2DYE PT-2DYE 2 12 16614664 16715489 100.825 DEL 11.82
S1 PT-264I PT-264I 2 12 16614664 16751045 136.381 DEL 14.2
S1 PT-25WZ PT-25WZ 1 12 16591338 16715767 124.429 DEL 14.7
S1 CON 14 8:6 12 16631570 16708856 77.286 NA NA
S1 UNION 14 8:6 12 16591338 17183201 591.863 NA NA
For CNV data (in contrast to shared segments based on homozygosity or IBD sharing) the
extra fields of TYPE (deletion or duplication) and SCORE (some metric
of quality/confidence of CNV call) are also presented.
Here we see the 14 segments listed, 8 cases and 6
controls. The CON and UNION lines at the end of the
pool give the consensus region (i.e. shared by all segments) and the
total distance spanned by all. The PHE field gives the
phenotype for each individual.
Note that the way in which the dummy markers are selected will effectively mean that every
possibly unique position, in terms of counts of segments, is evaluated. The actual base
pair regions of any dummy marker is itself probably not of interest: given a sigificant (set of)
SNPs, the strategy would be to select any one and generate the corresponding pool to see
what and where the association maps to.